联系我们
- 电话/微信:18520902353
- 客服QQ:3597831168
- 邮箱:info@wemaxnano.com
- 地址:广东省佛山市南海区狮山镇长兴西路12号建发风梅岭产业园9号楼

随着全球能源转型步伐的加速,氢能作为一种清洁、高效的二次能源,正逐渐成为替代化石燃料的重要选择。电解水制氢(Water Electrolysis)因其过程清洁、产物纯度高等优势,被视为实现绿氢规模化生产的关键技术之一。然而,电解水过程中的氧析出反应(OER) 因其四电子转移过程的缓慢动力学,成为制约整个水分解效率的瓶颈。
目前,商用OER催化剂主要依赖贵金属氧化物(如IrO₂、RuO₂),但其高昂的成本和稀缺性严重限制了大规模应用。因此,开发非贵金属、高效、稳定的OER催化剂已成为能源材料领域的研究热点。过渡金属硒化物(如CoSe)因具有良好的导电性和催化活性而受到关注,但其单金属组分往往面临活性位点单一、中间体吸附能调控能力有限、稳定性不足等问题。
近年来,高熵材料(HEMs) 因其多元素协同效应、独特的电子结构和优异的稳定性,在催化领域展现出巨大潜力。然而,如何实现多金属元素(尤其是互不相容元素)的均匀混合,并明确其活性位点的本质,仍是该领域面临的重大挑战。
中国科学院赣江创新研究院彭立山教授、中国科学技术大学宋礼教授和陈清军教授团队在《Joule》期刊发布了高熵金属硒化物催化剂(HEMS)的最新研究成果。该团队通过快速焦耳加热法成功合成了具有不对称活性位点的五元高熵金属硒化物。这一成果为高效稳定的氧析出反应催化剂设计提供了新策略,并为多金属协同催化机制提供了理论依据。

本研究成功开发了一种超快速焦耳加热(Ultrafast Joule Heating) 合成策略,首次实现了五元高熵金属硒化物(HEMS, (FeCoNiCrCu)Seₓ) 纳米颗粒的制备。该研究的创新性突破主要体现在以下三个方面:
此外,研究团队通过准原位X射线吸收光谱(XAFS)、操作衰减全反射红外光谱(operando ATR-IR) 以及理论计算(DFT) 等多种先进表征技术,首次从原子和电子层面揭示了高熵诱导的不对称活性位的形成机制及其增强OER性能的机理,为理性设计高性能高熵催化剂提供了深刻的理论见解和实践指导。
超快速焦耳热合成工艺:研究团队将五种金属盐(Fe, Co, Ni, Cr, Cu)与导电碳黑(Keijen black)均匀混合的前驱体与硒粉,在定制化装置中通过焦耳加热,在约20秒内经历超快升温和冷却过程(升温速率~400 °C/s),瞬间完成硒化反应,成功制备出一系列不同硒含量的(FeCoNiCrCu)Seₓ纳米颗粒。
结构与成分均匀性验证:
XRD与电子衍射(SAED):结果表明所有样品均为纯相的六方CoSe结构(PDF#89-2004),无其他杂相峰,证明了单相固溶体的形成。
(AC-)STEM与EDS mapping:原子分辨率STEM图像清晰显示了硒和金属原子的柱状排列。高角环形暗场像(HAADF-STEM)及面扫图谱直观地展示了所有五种金属元素和硒元素在纳米颗粒内呈原子级均匀分布,无任何元素偏聚或析出,有力证实了高熵态的成功实现。
卓越的OER活性:在1.0 M KOH电解液中,HEMS的性能远超单金属CoSe、前驱体合金(HEA)及商用IrO₂。
低过电位:仅需222 mV的过电位即可达到10 mA cm⁻²的电流密度,在100 mA cm⁻²时过电位为305 mV。
快速动力学:Tafel斜率低至55.5 mV dec⁻¹,接近IrO₂(58.5 mV dec⁻¹),表明反应动力学更快。
高本征活性:通过双电层电容(Cdl)和周转频率(TOF)计算证明,其卓越性能源于高本征活性,而非仅因比表面积增大。
卓越的稳定性:
长期稳定性:在10 mA cm⁻²和100 mA cm⁻²的电流密度下,分别持续运行1000小时和500小时,电位未见明显衰减。
工业级测试:在60°C的AEM电解槽中,以HEMS为阳极,在0.5 A cm⁻²的高电流密度下稳定运行100小时,展现了其在实际应用场景下的巨大潜力。
准原位XAFS:在不同外加电位下对HEMS进行测试,发现随着电位升高,Co、Ni等金属的K边吸收边向高能移动,表明其氧化态升高;FT-EXAFS中M-Se键峰强度增加且发生轻微位移,揭示了OER过程中活性位点局部配位环境的动态演变及氧物种的吸附。
操作ATR-IR:在1.5 V (vs. RHE) 电位下,于1020 cm⁻¹和1220 cm⁻¹处分别观察到归属于关键中间体O和OOH的特征吸收峰。HEMS上的信号强度显著强于CoSe,表明其对中间体的吸附能力更强,有利于加速反应动力学。
DFT理论计算:
确定了不对称的Cu-Co-Ni三原子单元是真正的活性中心。
自由能计算表明,HEMS上的速率决定步骤(RDS,即*OOH的形成)能垒显著低于CoSe上的对称Co位点。
d带中心理论和态密度(PDOS)分析表明,高熵效应优化了金属d电子结构,调控了与中间体的吸附强度,从而提升了本征活性。
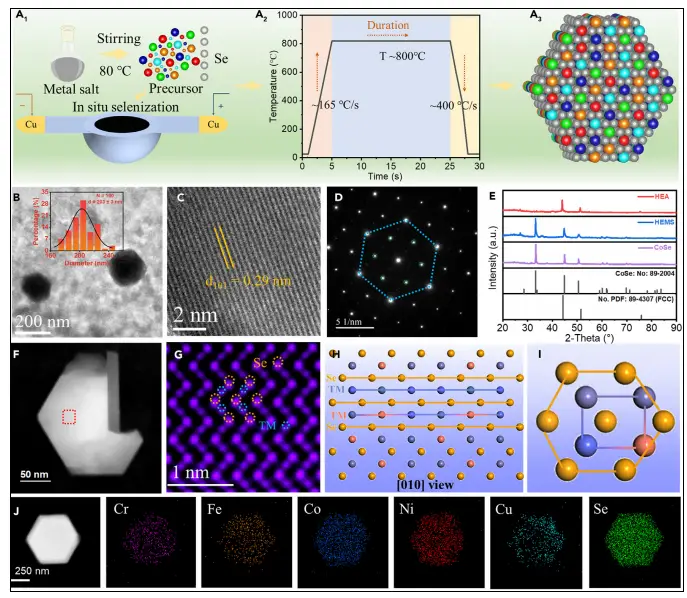
该图展示了通过快速焦耳加热法合成HEMS的工艺流程及其微观结构。图中包括前驱体混合、瞬时高温硒化过程,以及最终形成的均匀分散的五金属(Fe、Co、Ni、Cr、Cu)硒化物纳米颗粒。TEM和HRTEM图像显示HEMS具有约200 nm的多面体形貌和清晰的六方相晶格条纹,SAED图谱进一步证实其单相结构。EDS mapping显示各元素分布均匀,无偏析现象,表明成功实现了高熵结构的构建。
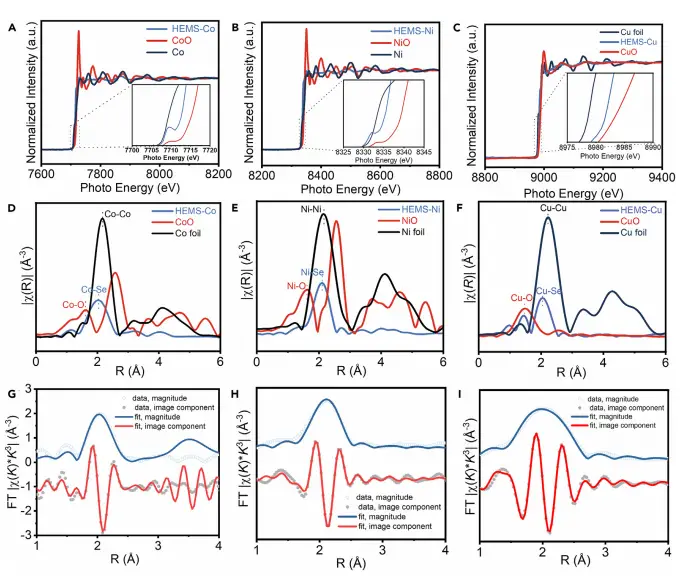
图2:HEMS中金属位点的配位环境分析
通过XANES和EXAFS技术,该图揭示了HEMS中Co、Ni、Cu等金属的局部电子结构和配位环境。XANES谱显示金属吸收边向高能区移动,表明其氧化态高于金属态;EXAFS拟合结果确认了金属与Se之间的键合作用,形成了稳定的TM-Se配位结构。这些结果共同表明,高熵原子的引入引发了电子重分布,为不对称活性位点的形成奠定了基础。
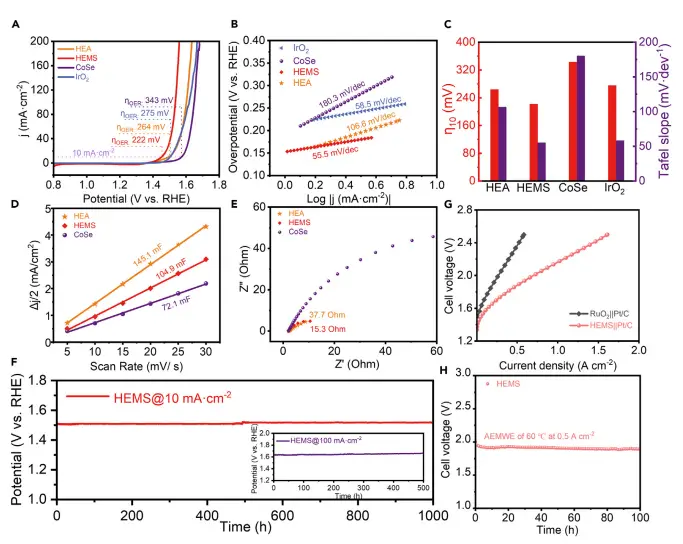
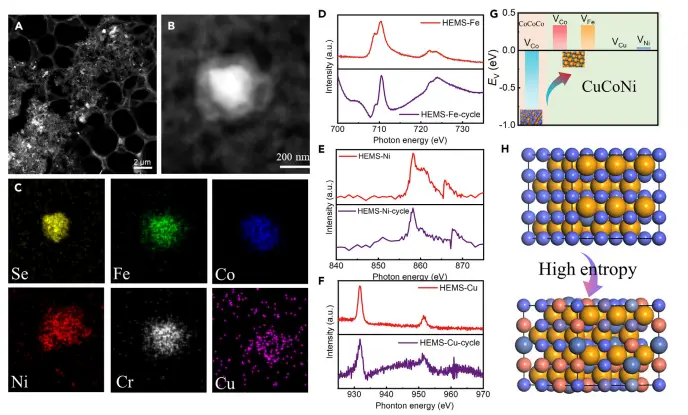
图4:HEMS的稳定性机制研究
通过对比HEMS在长期OER测试前后的结构变化,该图揭示了其高稳定性的内在机制。TEM图像显示尽管颗粒形貌略有变化,但各元素仍均匀分布,无明显的相分离或溶解。sXAS谱图显示Co、Ni、Cu的电子结构基本保持不变。DFT计算进一步表明,高熵结构的引入显著提高了表面金属原子的空位形成能,有效抑制了活性位点的流失。
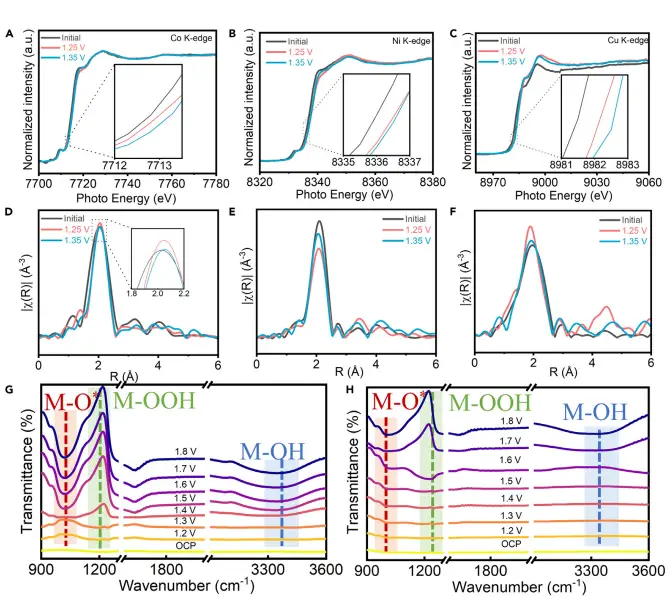
图5:准原位XAFS与操作ATR-IR揭示反应中间体行为
该图通过准原位XAFS和操作ATR-IR技术,实时监测了HEMS在OER过程中金属位点的电子结构变化和中间体吸附行为。随着电位升高,金属氧化态逐渐升高,O和OOH中间体的红外特征峰逐渐出现并增强,表明HEMS表面对氧中间体具有更强的吸附能力,从而促进了OER反应动力学。
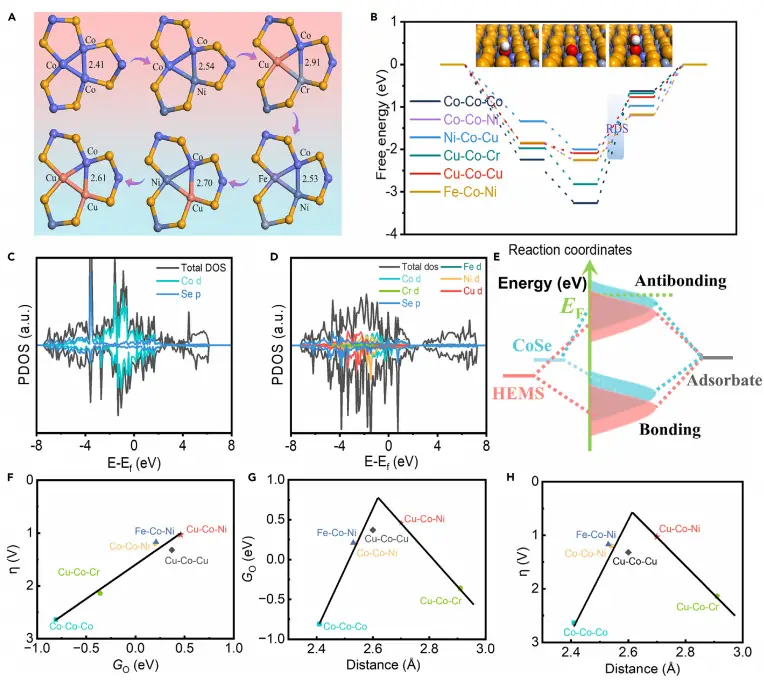
图6:DFT理论计算揭示不对称活性位点的作用机制
通过DFT计算,该图对比了对称Co-Co-Co位点与不对称Cu-Co-Ni位点在OER过程中的自由能变化。结果表明,不对称位点显著降低了*OOH形成的能垒(RDS),从而提升了本征催化活性。PDOS分析显示高熵结构增强了Se p轨道与金属d轨道的杂化,优化了中间体的吸附强度,进一步验证了不对称单元在提升OER性能中的关键作用。
本研究通过快速焦耳加热法成功合成了具有单一六方相结构的五元高熵金属硒化物(HEMS),并系统验证了其卓越的OER催化活性和稳定性。通过多种原位/准原位光谱技术和理论计算,首次揭示了其活性来源于不对称Cu-Co-Ni单元,该结构能有效调控中间体的吸附行为,降低反应能垒。
该工作不仅提供了一种高效、快速、可扩展的高熵材料合成策略,更为高熵催化剂的设计提供了新的视角:即通过多元素协同构建不对称活性中心,实现催化性能的突破。未来,该方法可进一步拓展至其他高熵硫族化合物(如硫化物、碲化物)或氮/磷化物,推动其在电解水、燃料电池、CO₂还原等能源转换技术中的实际应用。
文章来源:

联系客服二维码

纳米纤维及其应用
18520902353


info@wemaxnano.com